酮体的多维作用
酮体由肝脏产生,当葡萄糖在人体中不易获得时用作能量来源。 两种主要的酮体是乙酰乙酸酯 (AcAc) 和 3-β-羟基丁酸酯 (3HB),而丙酮是第三种也是最不丰富的酮体。 酮体始终存在于血液中,并且在禁食和长时间运动期间它们的水平会增加。生酮作用 是生物体通过分解脂肪酸和生酮氨基酸产生酮体的生化过程。
酮体主要产生于 肝细胞线粒体. 当血液中的葡萄糖水平低时,尤其是在其他细胞碳水化合物储存(如糖原)耗尽后,就会发生生酮作用。 当胰岛素量不足时,也会发生这种机制。 酮体的产生最终被启动以提供可用的能量,这些能量以脂肪酸的形式储存在人体内。 生酮发生在线粒体中,在那里它被独立调节。
抽象
酮体代谢是生理稳态的中心节点。 在这篇综述中,我们讨论了酮如何发挥离散的微调代谢作用,优化器官和生物体在不同营养残留物中的表现,并保护多个器官系统免受炎症和损伤。 传统上被视为仅在碳水化合物限制中加入的代谢底物,最近的观察强调了当碳水化合物丰富时酮体作为重要的代谢和信号传导介质的重要性。 作为对神经系统疾病已知治疗方案的补充,酮体在癌症中的预期作用已经出现,在心脏和肝脏中也具有有趣的保护作用,为肥胖相关疾病和心血管疾病开辟了治疗选择。 讨论了酮代谢和信号传导方面的争议,以调和经典教条与当代观察。
介绍
酮体是所有生命领域、真核生物、细菌和古细菌的重要替代代谢燃料来源(Aneja 等人,2002;Cahill GF Jr,2006;Krishnakumar 等人,2008)。 人类的酮体代谢已被用来在营养缺乏的偶发时期为大脑提供燃料。 酮体与关键的哺乳动物代谢途径交织在一起,例如 α-氧化 (FAO)、三羧酸循环 (TCA)、糖异生、从头脂肪生成 (DNL) 和甾醇的生物合成。 在哺乳动物中,酮体主要在肝脏中由FAO衍生的乙酰辅酶A产生,它们被运输到肝外组织进行终末氧化。 这种生理学提供了一种替代燃料,通过相对较短的禁食时间来增强,这增加了脂肪酸的利用率并减少了碳水化合物的利用率(Cahill GF Jr,2006;McGarry 和 Foster,1980;Robinson 和 Williamson,1980)。 在多种生理状态下,酮体氧化成为哺乳动物肝外组织内整体能量代谢的重要贡献者,包括禁食、饥饿、新生儿期、运动后、怀孕和坚持低碳水化合物饮食。 健康成年人的循环总酮体浓度通常表现出约 100-250 μM 之间的昼夜节律波动,在长时间运动或禁食 1 小时后升高至约 24 mM,并且在糖尿病酮症酸中毒等病理状态下可累积高达 20 mM。 Cahill GF Jr,2006;Johnson 等人,1969b;Koeslag 等人,1980;Robinson 和 Williamson,1980;Wildenhoff 等人,1974)。 人体肝脏每天可产生高达 300 克的酮体(Balasse 和 Fery,1989 年),占进食、禁食和饥饿状态下总能量消耗的 5-20%(Balasse 等人,1978 年;Cox 等人)等人,2016)。
最近的研究现在强调了酮体在哺乳动物细胞代谢、体内平衡和各种生理和病理状态下的信号传导中的重要作用。 除了作为脑、心脏或骨骼肌等肝外组织的能量燃料外,酮体还作为信号传导介质、蛋白质翻译后修饰 (PTM) 的驱动因素以及炎症和氧化应激的调节剂发挥着关键作用。 在这篇综述中,我们提供了关于酮体的多效作用及其代谢的经典和现代观点。
酮体代谢概述
肝脏生酮的速率受一系列精心安排的脂肪生理和生化转化的控制。 主要调节剂包括脂肪酸从三酰基甘油脂解、转运至肝细胞质膜并穿过肝细胞质膜、通过肉碱棕榈酰转移酶 1 (CPT1) 转运至线粒体、α-氧化螺旋、TCA 循环活性和中间浓度、氧化还原电位和激素调节剂在这些过程中,主要是胰高血糖素和胰岛素 [综述于 (Arias et al., 1995; Ayte et al., 1993; Ehara et al., 2015; Ferre et al., 1983; Kahn et al., 2005; McGarry and Foster ,1980 年;威廉姆森等人,1969 年)]。 经典的生酮被视为溢出途径,其中 α-氧化衍生的乙酰辅酶 A 超过柠檬酸合酶活性和/或草酰乙酸可用于缩合以形成柠檬酸。 三碳中间体表现出抗生酮活性,可能是由于它们能够扩大草酰乙酸池以消耗乙酰辅酶A,但仅肝脏乙酰辅酶A浓度并不能决定生酮率(Foster,1967;Rawat 和 Menahan,1975;Williamson等人,1969)。 激素、转录和翻译后事件对生酮的调节共同支持了微调生酮率的分子机制仍未完全了解的观点(参见 HMGCS2 和 SCOT/OXCT1 的调节)。
生酮主要以与总脂肪氧化成比例的速率发生在肝线粒体基质中。 在酰基链跨线粒体膜转运和 α-氧化后,3-羟甲基戊二酰辅酶 A 合酶 (HMGCS2) 的线粒体异构体催化乙酰乙酰辅酶 A (AcAc-CoA) 和乙酰辅酶 A 缩合生成 HMG-CoA (图 1A)。 HMG-CoA 裂解酶 (HMGCL) 裂解 HMG-CoA 以释放乙酰辅酶 A 和乙酰乙酸 (AcAc),后者被磷脂酰胆碱依赖性线粒体 d-αOHB 脱氢酶还原为 d-α-羟基丁酸 (d-αOHB)。 BDH1)在 NAD+/NADH 耦合的近平衡反应中(Bock 和 Fleischer,1975;LEHNINGER 等人,1960)。 BDH1 平衡常数有利于 d-?OHB 的产生,但 AcAc/d-?OHB 酮体的比率与线粒体 NAD+/NADH 比率成正比,因此 BDH1 氧化还原酶活性调节线粒体氧化还原电位(Krebs 等,1969;威廉姆森等人,1967)。 AcAc 也可以自发脱羧生成丙酮(Pedersen,1929),这是人类酮症酸中毒的甜味来源(即,总血清酮体 > ~7 mM;AcAc pKa 3.6,?OHB pKa 4.7)。 酮体通过线粒体内膜转运的机制尚不清楚,但 AcAc/d-αOHB 通过单羧酸转运蛋白(在哺乳动物中,MCT 1 和 2,也称为溶质载体 16A 家族成员 1 和7) 并在循环中运输到肝外组织进行终末氧化 (Cotter et al., 2011; Halestrap and Wilson, 2012; Halestrap, 2012; Hugo et al., 2012)。 循环酮体的浓度高于肝外组织中的浓度(Harrison 和 Long,1940),表明酮体沿浓度梯度向下输送。 MCT1 的功能丧失突变与酮症酸中毒的自发发作有关,表明在酮体输入中起关键作用。

� 除了可能将酮体转化为非氧化命运(见酮体的非氧化代谢命运)外,肝细胞缺乏代谢它们产生的酮体的能力。 由肝脏从头合成的酮体(i)在肝外组织的线粒体中分解代谢为乙酰辅酶A,乙酰辅酶A可用于TCA循环进行终末氧化(图1A),(ii)转向脂肪生成或甾醇合成途径(图 1B),或 (iii) 从尿中排出。 作为一种替代的高能燃料,酮体在心脏、骨骼肌和大脑中被强烈氧化(Balasse 和 Fery,1989;Bentourkia 等,2009;Owen 等,1967;Reichard 等,1974;Sultan,1988 )。 肝外线粒体 BDH1 催化 ?OHB 氧化的第一个反应,将其转化为 AcAc(LEHNINGER 等人,1960;Sandermann 等人,1986)。 与 BDH2 仅具有 20% 序列同一性的细胞质 d-?OHB-脱氢酶 (BDH1) 对酮体具有高 Km,并且还在铁稳态中发挥作用 (Davuluri et al., 2016; Guo et al., 2006) . 在肝外线粒体基质中,在由独特的哺乳动物 CoA 转移酶琥珀酰-CoA:3-氧代酸-CoA 转移酶(SCOT,CoA 转移酶;由 OXCT1 编码),通过近平衡反应。 AcAc-CoA 水解释放的自由能大于琥珀酰-CoA,有利于 AcAc 的形成。 因此,酮体氧化通量由于质量作用而发生:丰富的 AcAc 供应和通过柠檬酸合酶快速消耗的乙酰辅酶 A 有利于 SCOT 形成 AcAc-CoA(+ 琥珀酸)。 值得注意的是,与葡萄糖(己糖激酶)和脂肪酸(酰基辅酶 A 合成酶)相比,将酮体 (SCOT) 激活为可氧化形式不需要 ATP 的投入。 可逆的 AcAc-CoA 硫解酶反应 [由 ACAA2(编码称为 T1 或 CT 的酶)、ACAT1(编码 T2)、HADHA 或 HADHB 编码的四种线粒体硫解酶中的任何一种催化] 产生两个乙酰辅酶 A 分子,进入 TCA 循环(Hersh 和 Jencks,1967;Stern 等人,1956;Williamson 等人,1971)。 在酮状态(即总血清酮> 500 μM)期间,酮体成为能量消耗的重要贡献者,并在组织中迅速利用,直到发生吸收或氧化饱和(Balasse 等,1978;Balasse 和 Fery,1989 ;埃德蒙等人,1987 年)。 一小部分来源于肝脏的酮体可以很容易地在尿液中测量,肾脏的利用和重吸收率与循环浓度成正比(Goldstein,1987;Robinson 和 Williamson,1980)。 在高度酮症状态(血浆中 > 1 mM)期间,酮尿可作为酮症的半定量报告,尽管大多数尿酮体临床试验检测到 AcAc 而不是 ?OHB(Klocker 等,2013)。
生酮底物及其对肝细胞代谢的影响
生酮底物包括脂肪酸和氨基酸(图 1B)。 氨基酸的分解代谢,尤其是亮氨酸,在吸收后产生约 4% 的酮体(Thomas 等,1982)。 因此,产生酮体的乙酰辅酶A底物库主要来自脂肪酸,因为在碳水化合物供应减少的状态下,丙酮酸主要通过回补作用进入肝脏 TCA 循环,即 ATP 依赖性羧化为草酰乙酸 (OAA) 或苹果酸(MAL),而不是氧化脱羧成乙酰辅酶A(Jeoung 等人,2012;Magnusson 等人,1991;Merritt 等人,2011)。 在肝脏中,葡萄糖和丙酮酸对生酮的贡献可以忽略不计,即使丙酮酸脱羧为乙酰辅酶 A 的作用最大(Jeoung 等,2012)。
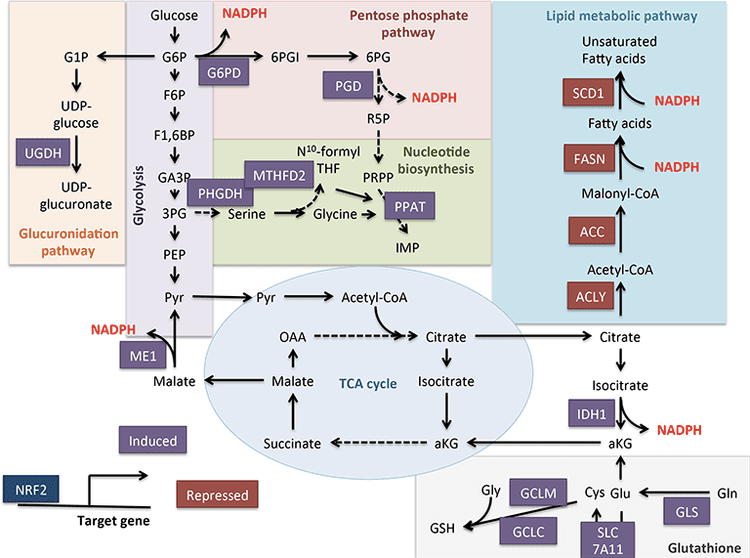
乙酰辅酶 A 在通过末端氧化产生 ATP 之外,还包含肝脏中间代谢不可或缺的几个角色(另见酮体代谢、翻译后修饰和细胞生理学的整合)。 乙酰辅酶 A 变构激活 (i) 丙酮酸羧化酶 (PC),从而激活代谢控制机制,增强代谢物回补进入 TCA 循环(Owen 等人,2002;Scruton 和 Utter,1967)和(ii)丙酮酸脱氢酶激酶,它磷酸化并抑制丙酮酸脱氢酶 (PDH) (Cooper et al., 1975),从而进一步增强丙酮酸通过回补作用流入 TCA 循环。 此外,细胞质乙酰辅酶A(其池通过将线粒体乙酰辅酶A转化为可转运代谢物的机制增加)抑制脂肪酸氧化:乙酰辅酶A羧化酶(ACC)催化乙酰辅酶A转化为丙二酰辅酶A,即脂肪生成底物和线粒体 CPT1 的变构抑制剂 [综述于 (Kahn et al., 2005; McGarry and Foster, 1980)]。 因此,线粒体乙酰辅酶A池既调节又受生酮溢出途径的调节,该途径协调肝脏中间代谢的关键方面。
酮体的非氧化代谢命运
肝源性酮的主要命运是 SCOT 依赖性肝外氧化。 然而,AcAc 可以从线粒体输出并通过由细胞质乙酰乙酰辅酶 A 合成酶催化的 ATP 依赖性反应转化为 AcAc-CoA 用于合成代谢途径(AACS,图 1B)。 该通路在大脑发育和哺乳期乳腺中是活跃的(Morris,2005;Robinson 和 Williamson,1978;Ohgami 等,2003)。 AACS 在脂肪组织和活化的破骨细胞中也高度表达(Aguilo 等人,2010;Yamasaki 等人,2016)。 细胞质 AcAc-CoA 可以被细胞质 HMGCS1 引导至甾醇生物合成,或被两种细胞质硫解酶中的任何一种裂解为乙酰辅酶 A(ACAA1 和 ACAT2),羧化为丙二酰辅酶 A,并有助于脂肪酸的合成(Bergstrom 等人)。 al., 1984; Edmond, 1974; Endemann et al., 1982; Geelen et al., 1983; Webber and Edmond, 1977)。
虽然生理意义尚未确定,但酮甚至可以在肝脏中作为合成代谢底物。 在人工实验环境中,AcAc 可贡献多达一半的新合成脂质和高达 75% 的新合成胆固醇(Endemann 等人,1982;Geelen 等人,1983;Freed 等人,1988)。 由于 AcAc 来源于不完全的肝脏脂肪氧化,因此 AcAc 在体内促进脂肪生成的能力意味着肝脏无效循环,其中脂肪衍生的酮可用于脂质生产,这一概念的生理意义需要实验验证,但可以服务适应或适应不良的角色(Solinas et al., 2015)。 AcAc 热衷于提供胆固醇生成,即使在进食状态下,AACS Km-AcAc (~50 µM) 也有利于 AcAc 活化(Bergstrom 等人,1984 年)。 细胞质酮代谢的动态作用已在原代小鼠胚胎神经元和 3T3-L1 衍生的脂肪细胞中提出,因为 AACS 敲低损害了每种细胞类型的分化(Hasegawa 等人,2012a;Hasegawa 等人,2012b)。 在体内敲除小鼠 AACS 可降低血清胆固醇(Hasegawa 等人,2012c)。 SREBP-2,胆固醇生物合成的主要转录调节因子,和过氧化物酶体增殖物激活受体 (PPAR)-? 是 AACS 转录激活剂,并在神经突发育和肝脏中调节其转录(Aguilo 等人,2010;Hasegawa 等人,2012c)。 总而言之,细胞质酮体代谢可能在特定条件或疾病自然史中很重要,但不足以处理源自肝脏的酮体,因为大量高酮血症发生在通过功能突变丧失对初级氧化命运的选择性损害的情况下到 SCOT(Berry 等人,2001 年;Cotter 等人,2011 年)。
HMGCS2和SCOT/OXCT1的调节
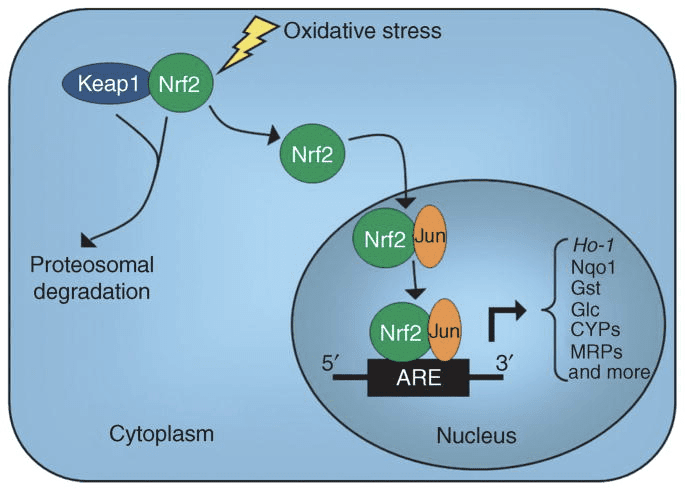
线粒体与编码细胞溶质 HMGCS 的基因的分歧发生在脊椎动物进化的早期,因为需要支持具有较高脑体重比的物种的肝生酮作用(Boukaftane 等,1994;Cunnane 和 Crawford,2003)。 人类自然发生的功能丧失 HMGCS2 突变会导致低酮症低血糖发作(Pitt 等人,2015;Thompson 等人,1997)。 强大的 HMGCS2 表达仅限于肝细胞和结肠上皮,其表达和酶活性通过多种机制进行协调(Mascaro 等,1995;McGarry 和 Foster,1980;Robinson 和 Williamson,1980)。 虽然影响 HMGCS2 的全部生理状态需要进一步阐明,但其表达和/或活性在产后早期、衰老、糖尿病、饥饿或摄入生酮饮食期间受到调节(Balasse 和 Fery,1989;Cahill GF Jr,2006 ;Girard 等人,1992;Hegardt,1999;Satapati 等人,2012;Sengupta 等人,2010)。 在胎儿中,Hmgcs5 基因 2’ 侧翼区域的甲基化与其转录呈负相关,并且在出生后部分逆转(Arias 等人,1995;Ayte 等人,1993;Ehara 等人,2015;Ferre 等人) ., 1983)。 类似地,肝脏 Bdh1 表现出发育表达模式,从出生到断奶增加,并且也由生酮饮食以成纤维细胞生长因子 (FGF)-21 依赖性方式诱导 (Badman et al., 2007; Zhang et al., 1989 )。 哺乳动物的生酮对胰岛素和胰高血糖素高度敏感,分别受到抑制和刺激(McGarry 和 Foster,1977)。 胰岛素抑制脂肪组织的脂肪分解,从而剥夺其底物的生酮作用,而胰高血糖素通过对肝脏的直接作用增加生酮通量(Hegardt,1999)。 Hmgcs2 转录受到叉头转录因子 FOXA2 的刺激,FOXA3 通过胰岛素-磷脂酰肌醇-300-激酶/Akt 抑制,并由胰高血糖素-cAMP-p1995 信号传导诱导(Arias 等,1999;Hegardt,1990;Quant 等。 , 1993; Thumelin 等人, 2013; von Meenn 等人, 2004; Wolfrum 等人, 2003; Wolfrum 等人, XNUMX)。 帕帕? (Rodriguez et al., 1994) 连同它的靶标 FGF21 (Badman et al., 2007) 也在饥饿或生酮饮食管理期间在肝脏中诱导 Hmgcs2 转录 (Badman et al., 2007; Inagaki et al., 2007) )。 PPAR的诱导? 可能发生在从胎儿到新生儿生理学的转变之前,而 FGF21 的激活可能通过 ?OHB 介导的组蛋白去乙酰化酶 (HDAC)-3 抑制在新生儿早期受到青睐 (Rando et al., 2016)。 mTORC1(雷帕霉素复合物 1 的哺乳动物靶标)对 PPAR 的依赖性抑制? 转录活性也是 Hmgcs2 基因表达的关键调节因子 (Sengupta et al., 2010),肝脏 PER2 是一种主要的昼夜节律振荡器,间接调节 Hmgcs2 表达 (Chavan et al., 2016)。 最近的观察表明,肝外肿瘤诱导的白细胞介素 6 通过 PPAR 损害酮生成? 抑制(Flint 等人,2016 年)。
HMGCS2 酶活性通过多个 PTM 进行调节。 HMGCS2 丝氨酸磷酸化增强了其体外活性(Grimsrud 等,2012)。 HMGCS2 活性受到琥珀酰辅酶 A 和赖氨酸残基琥珀酰化的变构抑制(Arias 等人,1995;Hegardt,1999;Lowe 和 Tubbs,1985;Quant 等人,1990;Rardin 等人,2013;Reed 等人, 1975 年;图梅林等人,1993 年)。 肝线粒体中 HMGCS2、HMGCL 和 BDH1 赖氨酸残基的琥珀酰化是 NAD+ 依赖性脱酰基酶 sirtuin 5 (SIRT5) 的靶标(Rardin 等,2013)。 SIRT2 赖氨酸脱乙酰化也增强了 HMGCS3 活性,乙酰化和琥珀酰化之间的串扰可能调节 HMGCS2 活性(Rardin 等人,2013;Shimazu 等人,2013)。 尽管这些 PTMs 能够调节 HMGCS2 Km 和 Vmax,但这些 PTMs 的波动尚未被仔细绘制,也未被证实为体内生酮的机制驱动因素。
SCOT 在所有含有线粒体的哺乳动物细胞中表达,肝细胞除外。 在 SCOT-KO 小鼠中证明了 SCOT 活性和酮分解的重要性,由于出生后 48 小时内出现高酮血症性低血糖,其表现出均匀的致死率(Cotter 等,2011)。 神经元或骨骼肌细胞中 SCOT 的组织特异性损失会在饥饿期间诱导代谢异常,但不是致命的(Cotter 等人,2013b)。 在人类中,SCOT 缺乏症在生命早期出现严重的酮症酸中毒,导致嗜睡、呕吐和昏迷(Berry 等人,2001;Fukao 等人,2000;Kassovska-Bratinova 等人,1996;Niezen-Koning 等人。 , 1997 年;Saudubray 等人,1987 年;Snyderman 等人,1998 年;Tildon 和 Cornblath,1972 年)。 在细胞水平上,关于 SCOT 基因和蛋白质表达调节剂的了解相对较少。 Oxct1 mRNA 表达和 SCOT 蛋白和活性在酮症状态下降低,可能通过 PPAR 依赖机制(Fenselau 和 Wallis,1974;Fenselau 和 Wallis,1976;Grinblat 等,1986;Okuda 等,1991;Turko 等.,2001 年;Wentz 等人,2010 年)。 在糖尿病酮症酸中毒中,肝脏生酮和肝外氧化之间的不匹配因SCOT活性受损而加剧。 心肌细胞中胰岛素非依赖性葡萄糖转运蛋白 (GLUT1/SLC2A1) 的过表达也会抑制 Oxct1 基因表达并下调非酮状态下的酮末端氧化 (Yan et al., 2009)。 在肝脏中,Oxct1 mRNA 丰度受到 microRNA-122 和组蛋白甲基化 H3K27me3 的抑制,这在从胎儿到新生儿期的过渡期间很明显(Thorrez 等,2011)。 然而,产后肝脏 Oxct1 表达的抑制主要是由于表达 Oxct1 的造血祖细胞从肝脏中排出,而不是终末分化肝细胞中先前存在的 Oxct1 表达的丧失。 事实上,Oxct1 mRNA 和 SCOT 蛋白在分化的肝细胞中的表达非常低(Orii 等,2008)。
SCOT 也受 PTM 监管。 该酶在 SIRT3 KO 小鼠的大脑中高度乙酰化,其也表现出 AcAc 依赖性乙酰辅酶 A 产生减少(Dittenhafer-Reed 等人,2015)。 SCOT 酪氨酸残基的非酶硝化也会减弱其活性,这已在各种糖尿病小鼠模型的心脏中得到报道(Marcondes 等人,2001;Turko 等人,2001;Wang 等人,2010a)。 相比之下,色氨酸残基硝化增强了 SCOT 活性(Br�g�re 等人,2010;Rebrin 等人,2007)。 旨在调节SCOT活性的残基特异性硝化或脱硝化的分子机制可能存在并且需要阐明。
肝外生酮的争议
在哺乳动物中,主要的生酮器官是肝脏,只有肝细胞和肠道上皮细胞大量表达 HMGCS2 的线粒体异构体(Cotter 等人,2013a;Cotter 等人,2014;McGarry 和 Foster,1980;Robinson 和 Williamson,1980) . 复杂多糖的厌氧细菌发酵产生丁酸盐,它被哺乳动物的结肠细胞吸收以进行终末氧化或生酮作用(Cherbuy 等人,1995 年),这可能在结肠细胞分化中发挥作用(Wang 等人,2016 年)。 除肠上皮细胞和肝细胞外,几乎所有其他哺乳动物细胞中几乎不存在 HMGCS2,但在肿瘤细胞、中枢神经系统的星形胶质细胞、肾脏、胰腺 ? 细胞、视网膜色素上皮 (RPE) 甚至骨骼肌(Adijanto 等人,2014;Avogaro 等人,1992;El Azzouny 等人,2016;Grabacka 等人,2016;Kang 等人,2015 ;Le Foll 等人,2014;Nonaka 等人,2016;Takagi 等人,2016a;Thevenet 等人,2016;Zhang 等人,2011)。 在缺乏净生酮能力的组织中观察到异位 HMGCS2 (Cook et al., 2016; Wentz et al., 2010),并且 HMGCS2 表现出预期的与生酮无关的“月光”活动,包括在细胞核内 (Chen et al., 2016)。 , 2010 年;Kostiuk 等人,1998 年;Meertens 等人,XNUMX 年)。
任何氧化酮体的肝外组织也有可能通过 HMGCS2 独立机制积累酮体(图 2A)。 然而,没有肝外组织的稳态酮体浓度超过循环中的浓度(Cotter 等人,2011;Cotter 等人,2013b;Harrison 和 Long,1940),这强调了酮体沿着通过 MCT1/2 依赖机制的浓度梯度。 一种明显的肝外生酮机制实际上可能反映了酮氧化的相对损害。 其他可能的解释属于酮体形成领域。 首先,从头生酮可能通过硫解酶和 SCOT 的可逆酶活性发生(Weidemann 和 Krebs,1969)。 当乙酰辅酶 A 的浓度相对较高时,通常负责 AcAc 氧化的反应会以相反的方向进行(GOLDMAN,1954)。 第二种机制发生在 α-氧化衍生的中间体由于 TCA 循环瓶颈而积累时,AcAc-CoA 通过线粒体 3-羟酰基-CoA 脱氢酶催化的反应转化为 L-βOHB-CoA,进一步由 3-羟基丁酰CoA 脱酰基酶生成 l-βOHB,通过质谱或共振光谱无法将其与生理对映异构体 d-βOHB 区分开来(Reed 和 Ozand,1980 年)。 l-?OHB 可以通过色谱或酶法与 d-?OHB 区分开来,并且存在于肝外组织中,但不存在于肝脏或血液中(Hsu 等,2011)。 肝脏生酮仅产生 d-βOHB,这是唯一一种 BDH 底物的对映异构体(Ito 等人,1984;Lincoln 等人,1987;Reed 和 Ozand,1980;Scofield 等人,1982;Scofield 等人, 1982)。 第三种不依赖 HMGCS2 的机制通过氨基酸分解代谢产生 d-βOHB,尤其是亮氨酸和赖氨酸的分解代谢。 第四种机制仅是显而易见的,因为它是由于标记伪影,因此被称为假酮生成。 这种现象归因于SCOT和硫解酶反应的可逆性,并且由于酮体示踪剂在肝外组织中的同位素稀释可能导致酮体周转率的高估(Des Rosiers et al., 1990; Fink et al., 1988) . 尽管如此,在大多数情况下,假酮生成可能可以忽略不计(Bailey 等人,1990;Keller 等人,1978)。 示意图(图 2A)表明了一种有用的方法来应用,同时考虑到酮的组织稳态浓度升高。


� 肾脏作为潜在的生酮器官最近受到关注。 在绝大多数州,肾脏是来自肝脏的酮体的净消耗者,从血液中排泄或再吸收酮体,而肾脏通常不是净酮体生成器或浓缩器(Robinson 和 Williamson,1980)。 一项经典研究的作者得出结论,在人工实验系统中量化的最小肾酮生成与生理学无关(Weidemann 和 Krebs,1969 年)。 最近,已在糖尿病和自噬缺陷小鼠模型中推断出肾生酮,但代谢稳态中的多器官变化更有可能通过多个器官的输入改变综合酮代谢(Takagi 等人,2016a;Takagi 等人, 2016b;张等人,2011)。 最近的一份出版物表明,肾生酮是一种针对肾脏缺血再灌注损伤的保护机制(Tran 等人,2016 年)。 据报道,小鼠肾组织提取物中 ?OHB 的绝对稳态浓度约为 4-12 mM。 为了测试这是否成立,我们量化了进食和 24 小时禁食小鼠肾脏提取物中的 ?OHB 浓度。 禁食 100 小时后,血清 ?OHB 浓度从约 2 μM 增加到 24 mM(图 2B),而肾稳态 ?OHB 浓度在进食状态下约为 100 μM,而在 1 小时禁食状态下仅为 24 mM(图 2)。 45C�E),与 1970 年前量化的浓度一致的观察结果(Hems 和 Brosnan,2014 年)。 在酮症状态下,肝脏来源的酮体可能具有肾脏保护作用,但肾脏生酮的证据需要进一步证实。 RPE 提供了支持真正肝外生酮的有力证据(Adijanto 等人,XNUMX 年)。 这种有趣的代谢转化被认为有可能使 RPE 衍生的酮流向光感受器或 Mller 神经胶质细胞,这可能有助于光感受器外段的再生。
?OHB 作为信号中介
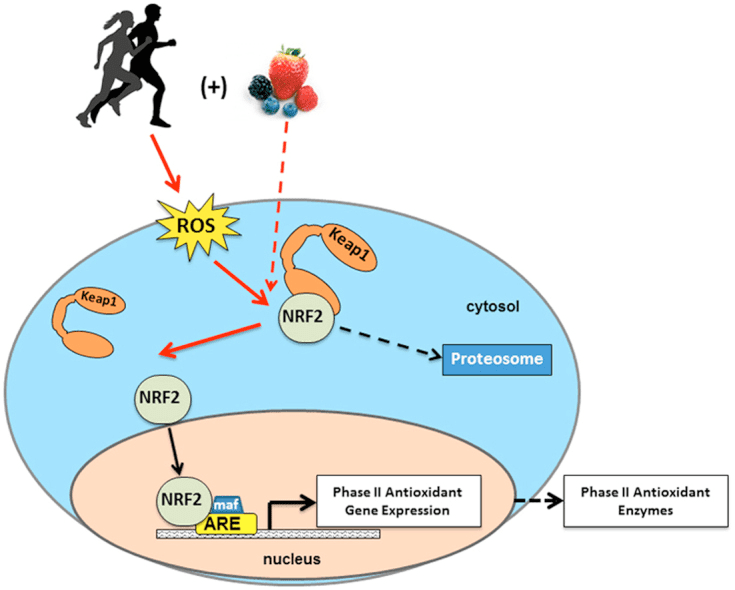
尽管它们能量丰富,但酮体在细胞稳态中发挥着挑衅性的“非典型”信号传导作用(图 3)(Newman 和 Verdin,2014;Rojas-Morales 等,2016)。 例如,?OHB 抑制 I 类 HDAC,增加组蛋白乙酰化,从而诱导抑制氧化应激的基因表达(Shimazu 等,2013)。 ?OHB 本身是禁食或链脲佐菌素诱导的糖尿病小鼠肝脏中赖氨酸残基的组蛋白共价修饰剂(Xie et al., 2016)(另见下文,酮体代谢、翻译后修饰和细胞生理学的整合,以及酮体、氧化应激和神经保护)。

?OHB 也是通过 G 蛋白偶联受体的效应器。 通过不清楚的分子机制,它通过 G 蛋白偶联受体 41 (GPR41) 抑制短链脂肪酸信号传导,从而抑制交感神经系统活动并降低总能量消耗和心率(Kimura 等,2011)。 ?OHB 研究最多的信号传导效应之一通过 GPR109A(也称为 HCAR2)进行,它是在脂肪组织(白色和棕色)中表达的羟基羧酸 GPCR 亚家族的成员(Tunaru 等,2003),并且在免疫细胞(Ahmed 等,2009)。 ?OHB 是唯一已知的 GPR109A 受体 (EC50 ~770 µM) 的内源性配体,可被 d-?OHB、l-?OHB 和丁酸盐激活,但不被 AcAc 激活 (Taggart et al., 2005)。 GPR109A 激活的高浓度阈值是通过坚持生酮饮食、饥饿或在酮症酸中毒期间实现的,从而抑制脂肪组织的脂肪分解。 GPR109A 的抗脂解作用通过抑制腺苷酸环化酶和降低 cAMP、抑制激素敏感性甘油三酯脂肪酶来进行(Ahmed 等人,2009;Tunaru 等人,2003)。 这产生了一个负反馈回路,其中酮症通过减少脂肪细胞中非酯化脂肪酸的释放来调节生酮作用(Ahmed et al., 2009; Taggart et al., 2005),这种效应可以通过以下方式抵消刺激脂肪分解的交感神经驱动。 烟酸(维生素 B3,烟酸)是 GRP50A 的有效 (EC0.1 ~ 109 μM) 配体,几十年来一直有效用于治疗血脂异常(Benyo 等人,2005;Benyo 等人,2006;Fabbrini 等人,2010a; Lukasova 等人,2011;Tunaru 等人,2003)。 虽然烟酸增强巨噬细胞中的反向胆固醇转运并减少动脉粥样硬化病变(Lukasova 等,2011),但 ?OHB 对动脉粥样硬化病变的影响仍然未知。 尽管 GPR109A 受体发挥保护作用,并且在中风和神经退行性疾病中使用生酮饮食之间存在有趣的联系(Fu 等人,2015;Rahman 等人,2014),但通过 GPR109A 的 ?OHB 的保护作用尚未在体内得到证实.
最后,?OHB 可能会影响食欲和饱腹感。 一项测量生酮饮食和极低能量饮食影响的研究的荟萃分析得出结论,与对照饮食相比,食用这些饮食的参与者表现出更高的饱腹感(Gibson 等人,2015 年)。 然而,对这种影响的一个合理解释是可能调节食欲的额外代谢或激素元素。 例如,与食物对照喂养的小鼠相比,维持啮齿动物生酮饮食的小鼠表现出增加的能量消耗,尽管卡路里摄入量相似,并且循环瘦素或调节摄食行为的肽基因没有改变(Kennedy 等,2007)。 建议的 ?OHB 抑制食欲的机制包括信号传导和氧化(Laeger et al., 2010)。 昼夜节律基因 (Per2) 的肝细胞特异性缺失和染色质免疫沉淀研究表明,PER2 直接激活 Cpt1a 基因,并间接调节 Hmgcs2,导致 Per2 敲除小鼠的酮症受损 (Chavan et al., 2016)。 这些小鼠表现出对食物的预期受损,通过全身性 ?OHB 给药部分恢复。 未来的研究将需要确认中枢神经系统是直接的?OHB 目标,以及观察到的效果是否需要酮氧化,或者是否涉及其他信号机制。 其他研究人员已经援引下丘脑腹内侧局部星形胶质细胞衍生的生酮作为食物摄入的调节剂的可能性,但这些初步观察也将受益于遗传和基于通量的评估(Le Foll 等,2014)。 酮症和营养缺乏之间的关系仍然很有趣,因为饥饿和饱腹感是减肥失败的重要因素。
酮体代谢、翻译后修饰和细胞生理学的整合
酮体有助于乙酰辅酶 A 的分隔池,这是一种在细胞代谢中表现出突出作用的关键中间体(Pietrocola 等人,2015 年)。 乙酰辅酶 A 的一个作用是作为乙酰化的底物,这是一种酶催化的组蛋白共价修饰(Choudhary 等人,2014;Dutta 等人,2016;Fan 等人,2015;Menzies 等人,2016 )。 计算蛋白质组学研究也出现了大量动态乙酰化线粒体蛋白,其中许多可能通过非酶机制发生(Dittenhafer-Reed 等人,2015;Hebert 等人,2013;Rardin 等人,2013) ;岛津等人,2010)。 赖氨酸脱乙酰酶使用锌辅助因子(例如,核细胞溶质 HDAC)或 NAD+ 作为共底物(sirtuins、SIRT)(Choudhary 等人,2014;Menzies 等人,2016)。 乙酰蛋白质组作为总细胞乙酰辅酶 A 池的传感器和效应器,因为生理和遗传操作都会导致乙酰化的非酶促全局变化(Weinert 等,2014)。 由于细胞内代谢物作为赖氨酸残基乙酰化的调节剂,重要的是要考虑酮体的作用,其丰度是高度动态的。
?OHB 是通过至少两种机制的表观遗传修饰剂。 禁食、热量限制、直接给药或长时间运动引起的 ?OHB 水平升高会引发 HDAC 抑制或组蛋白乙酰转移酶激活(Marosi 等人,2016;Sleiman 等人,2016)或氧化应激(Shimazu 等人,2013) . ?OHB 对 HDAC3 的抑制可以调节新生儿的代谢生理(Rando et al., 2016)。 独立地,?OHB 本身直接修饰组蛋白赖氨酸残基 (Xie et al., 2016)。 长时间禁食或后脲佐菌素诱导的糖尿病酮症酸中毒增加了组蛋白 α-羟基丁酰化。 尽管赖氨酸α-羟基丁酰化和乙酰化位点的数量相当,但在化学计量上观察到组蛋白α-羟基丁酰化比乙酰化更大。 与乙酰化或甲基化相比,组蛋白赖氨酸 α-羟基丁酰化影响不同的基因,表明不同的细胞功能。 α-羟基丁酰化是自发的还是酶促的尚不清楚,但通过酮体动态影响转录,扩大了机制的范围。
热量限制和营养剥夺期间的基本细胞重编程事件可能分别在 SIRT3 和 SIRT5 依赖性线粒体去乙酰化和去琥珀酰化中介导,在肝脏和肝外组织的翻译后水平调节生酮和酮分解蛋白(Dittenhafer-Reed 等人, 2015;Hebert 等人,2013;Rardin 等人,2013;Shimazu 等人,2010)。 尽管占据位点的化学计量比较不一定与代谢通量的变化直接相关,但线粒体乙酰化是动态的,可能由乙酰辅酶 A 浓度或线粒体 pH 值驱动,而不是酶促乙酰转移酶 (Wagner and Payne, 2013)。 SIRT3 和 SIRT5 调节酮体代谢酶的活性引发了酮在塑造乙酰蛋白质组、琥珀酰蛋白质组和其他动态细胞靶标中的相互作用的问题。 事实上,由于酮生成的变化反映了 NAD+ 浓度,酮的产生和丰度可以调节去乙酰化酶活性,从而影响总乙酰辅酶 A/琥珀酰辅酶 A 池、酰基蛋白质组,从而影响线粒体和细胞生理学。 赖氨酸残基的β-羟基丁酰化可以为细胞重编程增加另一层。 在肝外组织中,酮体氧化可能会刺激细胞稳态的类似变化。 虽然乙酰辅酶 A 池的分隔受到高度调节并协调广泛的细胞变化,但酮体直接塑造线粒体和细胞质乙酰辅酶 A 浓度的能力需要阐明(Chen 等人,2012;Corbet 等人, 2016;Pougovkina 等人,2014;Schwer 等人,2009;Wellen 和 Thompson,2012)。 由于乙酰辅酶 A 浓度受到严格调节,并且乙酰辅酶 A 是不透膜的,因此考虑协调乙酰辅酶 A 稳态的驱动机制至关重要,包括 TCA 循环中的生产和末端氧化速率、转化为酮体、线粒体通过肉毒碱乙酰转移酶 (CrAT) 流出,或乙酰辅酶 A 在转化为柠檬酸盐并通过 ATP 柠檬酸裂解酶 (ACLY) 释放后输出到细胞质。 这些后一种机制在细胞乙酰蛋白质组和体内平衡中的关键作用需要对生酮和酮氧化的作用有相匹配的理解(Das 等人,2015;McDonnell 等人,2016;Moussaieff 等人,2015;Overmyer 等人, 2015;Seiler 等人,2014;Seiler 等人,2015;Wellen 等人,2009;Wellen 和 Thompson,2012)。 在基因操作模型的设置中,代谢组学和酰基蛋白质组学的融合技术将需要指定目标和结果。
对酮体的抗炎和促炎反应
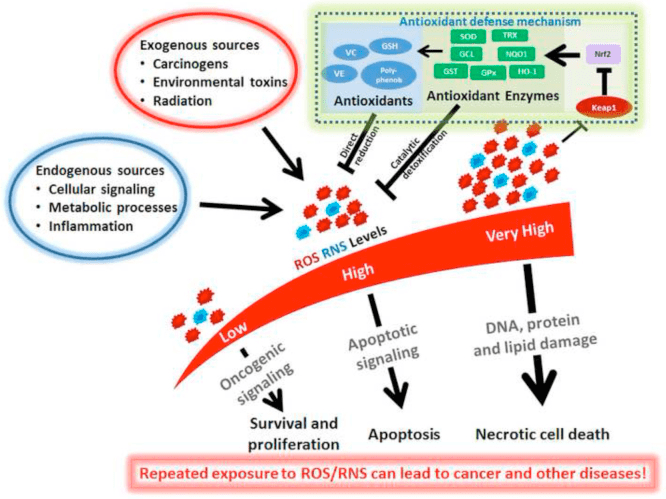
酮症和酮体调节炎症和免疫细胞功能,但已经提出了各种甚至不一致的机制。 长期缺乏营养会减少炎症(Youm 等,2015),但 1 型糖尿病的慢性酮症是一种促炎状态(Jain 等,2002;Kanikarla-Marie 和 Jain,2015;Kurepa 等,2012 )。 由于许多免疫系统细胞(包括巨噬细胞或单核细胞)大量表达 GPR109A,因此出现了 ?OHB 在炎症中基于机制的信号传导作用。 虽然 ?OHB 主要发挥抗炎反应(Fu 等人,2014;Gambhir 等人,2012;Rahman 等人,2014;Youm 等人,2015),但高浓度的酮体,尤其是 AcAc,可能引发促炎反应(Jain 等人,2002;Kanikarla-Marie 和 Jain,2015;Kurepa 等人,2012)。
GPR109A 配体在动脉粥样硬化、肥胖、炎症性肠病、神经系统疾病和癌症中的抗炎作用已被综述(Graff 等人,2016 年)。 GPR109A 在糖尿病模型、人类糖尿病患者(Gambhir 等人,2012 年)的 RPE 细胞和神经退行性变期间小胶质细胞中的表达增强(Fu 等人,2014 年)。 RPE 细胞中的 GPR109A 过表达增强了 ?OHB 的抗炎作用,并通过 GPR109A 的药理学抑制或基因敲除消除(Gambhir 等,2012)。 ?OHB 和外源性烟酸 (Taggart et al., 2005) 在 TNF? 中都具有抗炎作用。 或 LPS 通过降低促炎蛋白(iNOS、COX-2)或分泌细胞因子(TNFα、IL-1α、IL-6、CCL2/MCP-1)的水平,部分通过抑制 NF 诱导炎症-?B 易位(Fu 等人,2014;Gambhir 等人,2012)。 ?OHB 降低 ER 应激和 NLRP3 炎性体,激活抗氧化应激反应 (Bae et al., 2016; Youm et al., 2015)。 然而,在神经退行性炎症中,GPR109A 依赖性 ?OHB 介导的保护不涉及炎症介质,如 MAPK 通路信号传导(例如,ERK、JNK、p38)(Fu 等,2014),但可能需要 COX-1 依赖性 PGD2生产(Rahman 等人,2014 年)。 有趣的是,巨噬细胞 GPR109A 需要在缺血性中风模型中发挥神经保护作用(Rahman 等,2014),但 ?OHB 抑制骨髓来源巨噬细胞中 NLRP3 炎性体的能力与 GPR109A 无关(Youm 等., 2015)。 尽管大多数研究将 ?OHB 与抗炎作用联系起来,但 ?OHB 可能是促炎性的并增加小牛肝细胞中脂质过氧化的标志物(Shi 等,2014)。 因此,αOHB 的抗炎作用与促炎作用可能取决于细胞类型、βOHB 浓度、暴露持续时间以及共调节剂的存在与否。
与 ?OHB 不同,AcAc 可以激活促炎信号。 升高的 AcAc,尤其是高葡萄糖浓度时,通过 NADPH 氧化酶/氧化应激依赖性机制加剧内皮细胞损伤(Kanikarla-Marie 和 Jain,2015)。 糖尿病母亲脐带中的高 AcAc 浓度与较高的蛋白质氧化率和 MCP-1 浓度相关(Kurepa 等,2012)。 糖尿病患者的高 AcAc 与 TNF? 表达(Jain 等人,2002)和 AcAc,但不是 ?OHB,在 U1 人单核细胞中诱导 TNF?、MCP-937 表达、ROS 积累和降低的 cAMP 水平(Jain 等人,2002;Kurepa 等人., 2012)。
酮体依赖性信号现象通常仅在酮体浓度高(> 5 mM)时触发,并且在许多研究中,通过不清楚的机制将酮体与促炎或抗炎作用联系起来。 此外,由于 ?OHB 与 AcAc 对炎症的矛盾影响,以及 AcAc/?OHB 比率影响线粒体氧化还原电位的能力,评估酮体对细胞表型的作用的最佳实验比较了 AcAc 和 ? OHB 在不同的比率和不同的累积浓度下 [例如,(Saito et al., 2016)]。 最后,AcAc 只能作为锂盐或乙酯在商业上购买,需要在使用前进行碱水解。 锂阳离子独立诱导信号转导级联反应(Manji 等,1995),AcAc 阴离子不稳定。 最后,使用外消旋 d/l-?OHB 的研究可能会令人困惑,因为只有 d-?OHB 立体异构体可以被氧化成 AcAc,但 d-?OHB 和 l-?OHB 可以各自通过 GPR109A 发出信号,抑制 NLRP3 炎性体,并作为脂肪生成底物。
酮体、氧化应激和神经保护
氧化应激通常被定义为由于过度产生和/或消除受损而导致 ROS 过量呈现的状态。 酮体的抗氧化和氧化应激缓解作用已在体外和体内广泛描述,特别是在神经保护方面。 由于大多数神经元不能有效地从脂肪酸中产生高能磷酸盐,但在碳水化合物供应不足时会氧化酮体,因此酮体的神经保护作用尤为重要(Cahill GF Jr, 2006; Edmond et al., 1987; Yang等人,1987 年)。 在氧化应激模型中,BDH1 诱导和 SCOT 抑制表明酮体代谢可以重新编程以维持不同的细胞信号传导、氧化还原电位或代谢需求(Nagao 等人,2016;Tieu 等人,2003)。
酮体降低细胞损伤、损伤、死亡的等级,并降低神经元和心肌细胞的细胞凋亡(Haces 等人,2008;Maalouf 等人,2007;Nagao 等人,2016;Tieu 等人,2003)。 调用的机制是多种多样的,并不总是与浓度线性相关。 低毫摩尔浓度的 (d 或 l)-?OHB 清除 ROS(羟基阴离子),而 AcAc 清除许多 ROS 物种,但仅在浓度超过生理范围时(IC50 20-67 mM)(Haces 等人,2008 年) . 相反,对电子传输链的氧化还原电位的有益影响是一种通常与 d-?OHB 相关的机制。 虽然所有三种酮体(d/l-βOHB 和 AcAc)都减少了由糖酵解的化学抑制引发的神经元细胞死亡和 ROS 积累,但只有 d-βOHB 和 AcAc 阻止了神经元 ATP 下降。 相反,在体内低血糖模型中,(d 或 l)-αOHB 而不是 AcAc 可防止海马脂质过氧化(Haces 等人,2008;Maalouf 等人,2007;Marosi 等人,2016;Murphy,2009 ; Tieu 等人,2003 年)。 对喂食生酮饮食(87% 千卡脂肪和 13% 蛋白质)的小鼠进行的体内研究显示出抗氧化能力的神经解剖学变化(Ziegler 等,2003),其中海马体中观察到最深刻的变化,谷胱甘肽过氧化物酶和总抗氧化能力。
生酮饮食、酮酯(另见生酮饮食和外源性酮体的治疗用途)或 ?OHB 给药在缺血性中风模型中发挥神经保护作用(Rahman 等人,2014 年); 帕金森病(Tieu 等,2003); 中枢神经系统氧中毒发作(D'Agostino 等人,2013 年); 癫痫痉挛(Yum 等人,2015 年); 线粒体脑肌病、乳酸性酸中毒和中风样 (MELAS) 发作综合征(Frey 等人,2016 年)和阿尔茨海默病(Cunnane 和 Crawford,2003 年;Yin 等人,2016 年)。 相反,最近的一份报告证明了在线粒体 DNA 修复异常的转基因小鼠模型中,生酮饮食导致神经退行性进展的组织病理学证据,尽管线粒体生物合成和抗氧化特征有所增加(Lauritzen 等人,2016 年)。 其他相互矛盾的报告表明,暴露于高浓度酮体会引发氧化应激。 高剂量的 ?OHB 或 AcAc 会导致小牛肝细胞中的一氧化氮分泌、脂质过氧化、SOD、谷胱甘肽过氧化物酶和过氧化氢酶的表达降低,而在大鼠肝细胞中,MAPK 通路的诱导归因于 AcAc 而不是 ?OHB (Abdelmegeed et al., 2004) ; Shi et al., 2014; Shi et al., 2016)。
总之,大多数报告将 ?OHB 与氧化应激的减弱联系起来,因为它的给药抑制 ROS/超氧化物的产生,防止脂质过氧化和蛋白质氧化,增加抗氧化蛋白质水平,并改善线粒体呼吸和 ATP 产生(Abdelmegeed 等,2004; Haces 等人,2008;Jain 等人,1998;Jain 等人,2002;Kanikarla-Marie 和 Jain,2015;Maalouf 等人,2007;Maalouf 和 Rho,2008;Marosi 等人,2016;Tieu等人,2003;尹等人,2016;齐格勒等人,2003)。 虽然 AcAc 比 ?OHB 与氧化应激的诱导更直接相关,但这些影响并不总是容易从预期的促炎反应中分离出来(Jain 等,2002;Kanikarla-Marie 和 Jain,2015;Kanikarla-Marie 和耆那教,2016)。 此外,重要的是要考虑到多效性生酮饮食所赋予的明显抗氧化益处可能不会由酮体本身转导,并且酮体赋予的神经保护可能不完全归因于氧化应激。 例如,在葡萄糖剥夺期间,在皮质神经元葡萄糖剥夺模型中,?OHB 刺激自噬通量并阻止自噬体积累,这与减少神经元死亡有关(Camberos-Luna 等,2016)。 d-?OHB 还可通过抑制 HDAC 前瞻性地诱导经典抗氧化蛋白 FOXO3a、SOD、MnSOD 和过氧化氢酶(Nagao 等人,2016;Shimazu 等人,2013)。
非酒精性脂肪肝(NAFLD)和酮体代谢
肥胖相关的 NAFLD 和非酒精性脂肪性肝炎 (NASH) 是西方国家肝病的最常见原因 (Rinella 和 Sanyal, 2016),而 NASH 诱导的肝功能衰竭是肝移植最常见的原因之一。 虽然仅在肝细胞中过量储存超过 5% 肝重 (NAFL) 的三酰基甘油不会导致肝功能退化,但人类 NAFLD 的进展与全身胰岛素抵抗和 2 型糖尿病风险增加相关,并可能导致心血管疾病和慢性肾脏疾病(Fabbrini 等人,2009;Targher 等人,2010;Targher 和 Byrne,2013)。 NAFLD 和 NASH 的发病机制尚不完全清楚,但包括肝细胞代谢异常、肝细胞自噬和内质网应激、肝免疫细胞功能、脂肪组织炎症和全身炎症介质(Fabbrini 等,2009;Masuoka 和 Chalasani,2013 ;Targher 等人,2010 年;Yang 等人,2010 年)。 碳水化合物、脂质和氨基酸代谢的紊乱发生在人类和模型生物体中并导致肥胖、糖尿病和 NAFLD [综述于 (Farese 等人,2012;Lin 和 Accili,2011;Newgard,2012;Samuel 和舒尔曼,2012 年;孙和拉扎尔,2013 年)]。 虽然在 NAFLD 中通常观察到细胞质脂质代谢中的肝细胞异常(Fabbrini 等,2010b),但在 NAFLD 发病机制中控制脂肪氧化处理的线粒体代谢的作用尚不清楚。 线粒体代谢异常发生在 NAFLD/NASH 发病机制中并促成了 NAFLD/NASH 发病机制(Hyotylainen 等人,2016 年;Serviddio 等人,2011 年;Serviddio 等人,2008 年;Wei 等人,2008 年)。 有一般性(Felig et al., 1974; Iozzo et al., 2010; Koliaki et al., 2015; Satapati et al., 2015; Satapati et al., 2012; Sunny et al., 2011)但不统一( Koliaki 和 Roden,2013 年;Perry 等人,2016 年;Rector 等人,2010 年)一致认为,在真正的 NASH 发展之前,肝线粒体氧化,特别是脂肪氧化,在肥胖、全身性胰岛素抵抗中增强和 NAFLD。 很可能随着 NAFLD 的进展,甚至在单个线粒体中出现氧化能力异质性,最终氧化功能受损(Koliaki 等人,2015;Rector 等人,2010;Satapati 等人,2008;Satapati 等人) ., 2012)。
生酮通常被用作肝脏脂肪氧化的代表。 随着 NAFLD 在动物模型中的进展,酮生成的损害出现,并且可能在人类中出现。 通过不完全确定的机制,高胰岛素血症抑制生酮,与瘦对照相比可能导致低酮血症(Bergman 等人,2007;Bickerton 等人,2008;Satapati 等人,2012;Soeters 等人,2009;Sunny 等人。 ,2011 年;Vice 等人,2005 年)。 尽管如此,循环酮体浓度预测 NAFLD 的能力仍存在争议(M�nnist�等人,2015;Sanyal 等人,2001)。 动物模型中稳健的定量磁共振波谱方法显示,中度胰岛素抵抗时酮转换率增加,但胰岛素抵抗更严重时,酮转换率明显降低(Satapati 等人,2012 年;Sunny 等人,2010 年)。 在患有脂肪肝的肥胖人群中,生酮率是正常的(Bickerton 等人,2008;Sunny 等人,2011),因此,相对于肝细胞内脂肪酸负荷的增加,生酮率降低。 因此,α-氧化衍生的乙酰辅酶A 可能被导向 TCA 循环中的末端氧化、增加末端氧化、磷酸烯醇式丙酮酸驱动的糖异生通过回补/催化反应和氧化应激。 乙酰辅酶 A 也可能以柠檬酸盐的形式从线粒体输出,柠檬酸盐是脂肪生成的前体底物(图 4)(Satapati 等人,2015;Satapati 等人,2012;Solinas 等人,2015)。 虽然酮生成对胰岛素或长期肥胖的禁食反应降低(Satapati 等,2012),但其潜在机制和下游后果仍未完全了解。 最近的证据表明,mTORC1 以一种可能位于胰岛素信号下游的方式抑制酮生成(Kucejova 等人,2016),这与 mTORC1 抑制 PPAR? 介导的 Hmgcs2 诱导的观察结果一致(Sengupta 等人,2010)(另见 HMGCS2 和 SCOT/OXCT1 的调节)。

我们小组的初步观察表明生酮不足会对肝脏产生不利影响(Cotter 等人,2014 年)。 为了验证生酮受损的假设,即使在碳水化合物充足和“非生酮”状态下,也会导致葡萄糖代谢异常并引发脂肪性肝炎,我们通过给予反义寡核苷酸 (ASO) Hmgcs2。 标准低脂食物喂养的成年小鼠中 HMGCS2 的损失导致轻度高血糖症并显着增加数百种肝脏代谢物的产生,其中一组强烈表明脂肪生成激活。 对酮生成不足的小鼠进行高脂饮食喂养会导致广泛的肝细胞损伤和炎症。 这些发现支持以下中心假设:(i)生酮不是被动溢出途径,而是肝脏和综合生理稳态的动态节点,(ii)谨慎的生酮增强以减轻 NAFLD/NASH 和肝脏葡萄糖代谢紊乱值得探索.
生酮受损如何导致肝损伤和葡萄糖稳态改变? 首先要考虑的罪魁祸首是生酮通量不足,还是酮体本身。 最近的一份报告表明,酮体可以减轻对 n-3 多不饱和脂肪酸的反应而引起的氧化应激引起的肝损伤(Pawlak 等人,2015 年)。 回想一下,由于肝细胞中缺乏SCOT表达,酮体没有被氧化,但它们可以促进脂肪生成,并发挥多种信号作用,而与它们的氧化无关(另见酮体和 ?OHB 的非氧化代谢命运)信号中介)。 肝细胞衍生的酮体也可能作为肝腺泡内相邻细胞类型的信号和/或代谢物,包括星状细胞和枯否细胞巨噬细胞。 虽然可用的有限文献表明巨噬细胞不能氧化酮体,但这仅使用经典方法测量,并且仅在腹膜巨噬细胞中进行测量(Newsholme 等,1986;Newsholme 等,1987),表明重新考虑到骨髓来源的巨噬细胞中丰富的 SCOT 表达,评估是适当的(Youm 等人,2015)。
肝细胞生酮通量也可能具有细胞保护作用。 虽然有益的机制可能不依赖于生酮本身,但低碳水化合物生酮饮食与 NAFLD 的改善有关(Browning 等,2011;Foster 等,2010;Kani 等,2014;Schugar 和 Crawford,2012) . 我们的观察表明,肝细胞生酮可以反馈和调节 TCA 循环通量、回补通量、磷酸烯醇丙酮酸衍生的糖异生(Cotter 等人,2014),甚至糖原转换。 生酮损伤会导致乙酰辅酶 A 增加 TCA 通量,这与肝脏中 ROS 介导的损伤增加有关(Satapati 等人,2015;Satapati 等人,2012); 迫使碳转化为可以证明具有细胞毒性的从头合成的脂质种类; 并防止 NADH 再氧化为 NAD+(Cotter 等人,2014 年)(图 4)。 总之,未来的实验需要解决相对生酮不足可能变得适应不良、导致高血糖、引发脂肪性肝炎的机制,以及这些机制在人类 NAFLD/NASH 中是否有效。 流行病学证据表明,在脂肪性肝炎的进展过程中,酮生成受损(Embade 等人,2016;Marinou 等人,2011;M�nnist� 等人,2015;Pramfalk 等人,2015;Safaei 等人,2016)增加肝脏生酮的疗法可能是有益的(Degirolamo 等人,2016 年;本田等人,2016 年)。
酮体和心力衰竭 (HF)
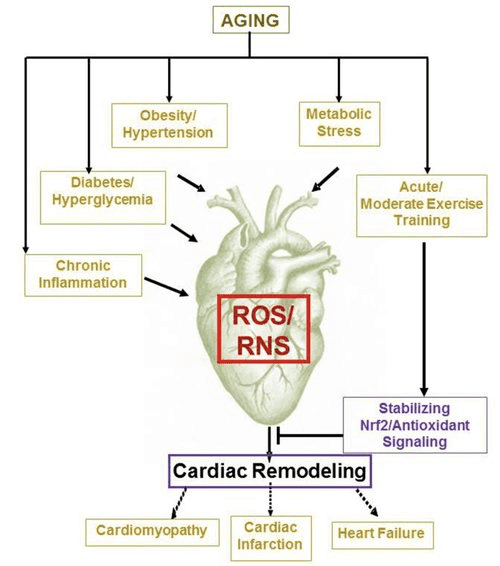
心脏代谢率超过 400 kcal/kg/天,ATP 周转率为 6~35 kg/天,是能量消耗和氧化需求最高的器官(Ashrafian et al., 2007; Wang et al., 2010b)。 绝大多数心肌能量周转位于线粒体内,其中 70% 的供应来自粮农组织。 在正常情况下,心脏是杂食性的和灵活的,但病理性重塑的心脏(例如,由于高血压或心肌梗塞)和糖尿病心脏都变得代谢不灵活(Balasse 和 Fery,1989;BING,1954;Fukao 等,2004 ;Lopaschuk 等人,2010;Taegtmeyer 等人,1980;Taegtmeyer 等人,2002;Young 等人,2002)。 事实上,小鼠模型中心脏燃料代谢的基因程序异常会引发心肌病(Carley 等人,2014;Neubauer,2007)。 在生理条件下,正常心脏按其输送的比例氧化酮体,以牺牲脂肪酸和葡萄糖氧化为代价,并且心肌是每单位质量的最高酮体消耗者(BING,1954;Crawford 等,2009;GARLAND 等., 1962; Hasselbaink 等人, 2003; Jeffrey 等人, 1995; Pelletier 等人, 2007; Tardif 等人, 2001; Yan 等人, 2009)。 与脂肪酸氧化相比,酮体的能量效率更高,每投入一分子氧(P/O 比)可产生更多可用于 ATP 合成的能量(Kashiwaya 等人,2010;Sato 等人,1995;Veech,2004) . 酮体氧化也可能产生比FAO更高的能量,保持泛醌被氧化,这提高了电子传递链中的氧化还原跨度,并为合成ATP提供了更多的能量(Sato et al., 1995; Veech, 2004)。 酮体的氧化也可能减少 ROS 的产生,从而减少氧化应激 (Veech, 2004)。
初步的介入性和观察性研究表明酮体在心脏中具有潜在的有益作用。 在实验性缺血/再灌注损伤背景下,酮体具有潜在的心脏保护作用(Al-Zaid 等,2007;Wang 等,2008),可能是由于心脏中线粒体丰度的增加或关键氧化磷酸化的上调调解员(Snorek 等人,2012 年;Zou 等人,2002 年)。 最近的研究表明,在小鼠(Aubert 等,2016)和人类(Bedi 等,2016)的心脏衰竭中,酮体的利用率增加,这支持了先前在人类中的观察结果(BING,1954;Fukao 等,2000; Janardhan 等人,2011;Longo 等人,2004;Rudolph 和 Schinz,1973;Tildon 和 Cornblath,1972)。 心力衰竭患者的循环酮体浓度增加,与充盈压力成正比,观察结果的机制和意义仍然未知(Kupari 等人,1995;Lommi 等人,1996;Lommi 等人,1997;Neely 等人) ., 1972), 但心肌细胞选择性 SCOT 缺乏的小鼠表现出加速的病理性心室重构和 ROS 特征,以响应手术引起的压力过载损伤 (Schugar et al., 2014)。
最近在糖尿病治疗中的有趣观察揭示了心肌酮代谢和病理性心室重塑之间的潜在联系(图 5)。 抑制肾近端肾小管钠/葡萄糖协同转运蛋白 2 (SGLT2i) 通过增加循环酮体浓度增加人类(Ferrannini 等人,2016a;Inagaki 等人,2015)和小鼠(Suzuki 等人,2014)肝脏生酮(Ferrannini 等人,2014;Ferrannini 等人,2016a;Katz 和 Leiter,2015;Mudaliar 等人,2015)。 引人注目的是,这些药物中至少有一种降低了 HF 住院率(例如,如 EMPA-REG OUTCOME 试验所揭示的),并提高了心血管死亡率(Fitchett 等人,2016;Sonesson 等人,2016;Wu 等人,2016a ;Zinman 等人,2015 年)。 虽然有益的 HF 结果与关联的 SGLT2i 背后的驱动机制仍然存在激烈争论,但生存益处可能是多因素的,预期包括酮症,但也对体重、血压、葡萄糖和尿酸水平、动脉僵硬度、交感神经系统、渗透利尿/减少血浆容量,增加血细胞比容(Raz 和 Cahn,2016;Vallon 和 Thomson,2016)。 总而言之,治疗性增加 HF 患者或发生 HF 高风险的酮血症的概念仍然存在争议,但正在临床前和临床研究中进行积极调查(Ferrannini 等人,2016b;Kolwicz 等人, 2016;Lopaschuk 和 Verma,2016;Mudaliar 等人,2016;Taegtmeyer,2016)。

癌症生物学中的酮体
酮体与癌症之间的联系正在迅速显现,但对动物模型和人类的研究得出了不同的结论。 由于酮代谢是动态的并且对营养状态有反应,因此追求与癌症的生物学联系是诱人的,因为有可能进行精确指导的营养疗法。 癌细胞经历代谢重编程以维持快速的细胞增殖和生长(DeNicola 和 Cantley,2015;Pavlova 和 Thompson,2016)。 癌细胞代谢中的经典 Warburg 效应源于糖酵解和乳酸发酵在传递能量和补偿对氧化磷酸化的较低依赖性和有限的线粒体呼吸方面的主要作用(De Feyter 等人,2016;Grabacka 等人,2016; Kang 等人,2015;Poff 等人,2014;Shukla 等人,2014)。 葡萄糖碳主要通过糖酵解、戊糖磷酸途径和脂肪生成,它们共同提供肿瘤生物量扩增所需的中间体(Grabacka 等人,2016;Shukla 等人,2014;Yoshii 等人,2015)。 癌细胞通过利用替代燃料来源(包括醋酸盐、谷氨酰胺和天冬氨酸)的能力来适应葡萄糖剥夺(Jaworski et al., 2016; Sullivan et al., 2015)。 例如,限制获取丙酮酸揭示了癌细胞通过羧化将谷氨酰胺转化为乙酰辅酶A的能力,从而维持能量和合成代谢的需求(Yang et al., 2014)。 癌细胞的一个有趣的适应是利用醋酸盐作为燃料(Comerford 等人,2014;Jaworski 等人,2016;Mashimo 等人,2014;Wright 和 Simone,2016;Yoshii 等人,2015)。 醋酸盐也是脂肪生成的底物,这对肿瘤细胞增殖至关重要,并且这种脂肪生成管道的获得与更短的患者生存期和更大的肿瘤负荷相关(Comerford 等人,2014 年;Mashimo 等人,2014 年;Yoshii 等人) ., 2015)。
在葡萄糖剥夺期间,非癌细胞很容易将其能量来源从葡萄糖转移到酮体。 这种可塑性在癌细胞类型中可能更具可变性,但体内植入的脑肿瘤将 [2,4-13C2]-?OHB 氧化到与周围脑组织相似的程度(De Feyter 等,2016)。 “反向 Warburg 效应”或“两室肿瘤代谢”模型假设癌细胞会在相邻的成纤维细胞中诱导 ?OHB 产生,从而满足肿瘤细胞的能量需求(Bonuccelli 等人,2010;Martinez-Outschoorn 等人,2012) . 在肝脏中,肝细胞癌 (hepatoma) 细胞中肝细胞从酮生成向酮氧化的转变与在两种肝癌细胞系中观察到的 BDH1 和 SCOT 活性的激活一致 (Zhang et al., 1989)。 事实上,肝癌细胞表达 OXCT1 和 BDH1 并氧化酮,但仅在血清饥饿时(Huang et al., 2016)。 或者,还提出了肿瘤细胞生酮作用。 生酮基因表达的动态变化表现在结肠上皮的癌变过程中,结肠上皮是一种通常表达 HMGCS2 的细胞类型,最近的一份报告表明 HMGCS2 可能是结直肠癌和鳞状细胞癌预后不良的预后标志物(Camarero 等人, 2006;陈等人,2016)。 这种关联是否需要或涉及生酮,或 HMGCS2 的兼职功能,仍有待确定。 相反,由 PPAR 刺激的黑色素瘤和胶质母细胞瘤细胞明显产生 ?OHB? 激动剂非诺贝特与生长停滞有关(Grabacka 等,2016)。 需要进一步的研究来表征 HMGCS2/SCOT 表达、生酮和酮氧化在癌细胞中的作用。
除了燃料代谢领域,酮最近还通过信号机制与癌细胞生物学有关。 对 BRAF-V600E+ 黑色素瘤的分析表明 OCT1 依赖性诱导 HMGCL 以致癌 BRAF 依赖性方式(Kang 等人,2015)。 HMGCL 增强与更高的细胞 AcAc 浓度相关,这反过来又增强了 BRAFV600E-MEK1 相互作用,在驱动肿瘤细胞增殖和生长的前馈回路中放大 MEK-ERK 信号。 这些观察提出了一个有趣的问题,即预期的肝外生酮,然后支持信号机制(另见 ?OHB 作为信号介质和肝外生酮的争议)。 考虑 AcAc、d-βOHB 和 l-βOHB 对癌症代谢的独立影响也很重要,当考虑 HMGCL 时,亮氨酸分解代谢也可能出现紊乱。
生酮饮食(另见生酮饮食和外源性酮体的治疗用途)在癌症动物模型中的作用各不相同(De Feyter 等人,2016;Klement 等人,2016;Meidenbauer 等人,2015;Poff 等人) .,2014 年;Seyfried 等人,2011 年;Shukla 等人,2014 年)。 虽然肥胖、癌症和生酮饮食之间的流行病学关联存在争议(Liskiewicz 等,2016;Wright 和 Simone,2016),但在动物模型和人体研究中使用生酮饮食的荟萃分析表明,生酮饮食对生存有有益的影响,益处与酮症程度、饮食开始时间和肿瘤位置前瞻性相关(Klement et al., 2016; Woolf et al., 2016)。 用酮体(d-βOHB 或 AcAc)处理胰腺癌细胞可抑制生长、增殖和糖酵解,生酮饮食(81% kcal 脂肪、18% 蛋白质、1% 碳水化合物)可降低体内肿瘤重量、血糖和植入癌症动物的肌肉和体重增加(Shukla 等,2014)。 在饮食中补充酮的小鼠中使用转移性胶质母细胞瘤细胞模型观察到了类似的结果(Poff 等,2014)。 相反,生酮饮食(91% kcal 脂肪,9% 蛋白质)增加循环 ?OHB 浓度并降低血糖,但对荷胶质瘤大鼠的肿瘤体积或存活时间没有影响(De Feyter 等,2016)。 已提出葡萄糖酮指数作为临床指标,可改善人类和小鼠生酮饮食诱导的脑癌治疗的代谢管理(Meidenbauer 等,2015)。 总而言之,酮体代谢和酮体在癌症生物学中的作用是诱人的,因为它们都提供了易于处理的治疗选择,但基本方面仍有待阐明,明确的影响来自变量矩阵,包括 (i) 外源酮之间的差异身体与生酮饮食,(ii)癌细胞类型、基因组多态性、等级和阶段; (iii) 暴露于酮症状态的时间和持续时间。

酮生成是由酮体通过脂肪酸和生酮氨基酸的分解而产生的。 这种生化过程在禁食的情况下为各种器官,特别是大脑提供能量,作为对血糖不可用的反应。 酮体主要在肝细胞的线粒体中产生。 虽然其他细胞能够进行生酮作用,但它们在这样做方面不如肝细胞有效。 因为生酮发生在线粒体中,其过程是独立调节的。 CCST Insight的Alex Jimenez博士
生酮饮食和外源性酮体的治疗应用
生酮饮食和酮体作为治疗工具的应用也出现在非癌症环境中,包括肥胖和 NAFLD/NASH(Browning 等人,2011;Foster 等人,2010;Schugar 和 Crawford,2012); 心力衰竭(Huynh,2016;Kolwicz 等人,2016;Taegtmeyer,2016); 神经和神经退行性疾病(Martin 等人,2016;McNally 和 Hartman,2012;Rho,2015;Rogawski 等人,2016;Yang 和 Cheng,2010;Yao 等人,2011); 先天性代谢错误(Scholl-B�rgi 等,2015); 和运动表现(Cox 等人,2016 年)。 生酮饮食的功效在癫痫发作的治疗中尤其受到重视,特别是在耐药患者中。 大多数研究已经评估了儿科患者的生酮饮食,并显示 50 个月后癫痫发作频率降低了约 3%,并提高了对特定综合征的有效性(Wu 等人,2016b)。 成人癫痫的经验更为有限,但类似的减少是明显的,对有症状的全身性癫痫患者有更好的反应(Nei et al., 2014)。 潜在的抗惊厥作用机制仍不清楚,尽管假设包括葡萄糖利用/糖酵解减少、谷氨酸转运重编程、对 ATP 敏感性钾通道或腺苷 A1 受体的间接影响、钠通道异构体表达的改变或对包括瘦素在内的循环激素的影响。 Lambrechts 等人,2016;Lin 等人,2017;Lutas 和 Yellen,2013)。 目前尚不清楚抗惊厥作用是主要归因于酮体,还是归因于低碳水化合物饮食的级联代谢后果。 尽管如此,酮酯(见下文)似乎提高了诱发癫痫发作动物模型的癫痫发作阈值(Ciarlone 等人,2016;D'Agostino 等人,2013;Viggiano 等人,2015)。
阿特金斯式和生酮低碳水化合物饮食通常被认为令人不快,并可能导致便秘、高尿酸血症、低钙血症、低镁血症、导致肾结石、酮症酸中毒、引起高血糖并提高循环胆固醇和游离脂肪酸浓度(Bisschop 等,2001 ;Kossoff 和 Hartman,2012 年;Kwiterovich 等人,2003 年;铃木等人,2002 年)。 由于这些原因,长期坚持会带来挑战。 啮齿动物研究通常使用独特的常量营养素分布(94% kcal 脂肪、1% kcal 碳水化合物、5% kcal 蛋白质、Bio-Serv F3666),这会引发强烈的酮症。 然而,增加蛋白质含量,即使是 10% kcal 也会大大减少酮症,而 5% kcal 的蛋白质限制会带来混杂的代谢和生理效应。 这种饮食配方也缺乏胆碱,这是另一个影响肝损伤易感性甚至生酮的变量(Garbow 等人,2011;Jornayvaz 等人,2010;Kennedy 等人,2007;Pissios 等人,2013;Schugar等人,2013)。 长期食用生酮饮食对小鼠的影响仍未完全确定,但最近对小鼠的研究表明,尽管氨基酸代谢、能量消耗和胰岛素信号转导,生酮饮食的小鼠在其一生中存活率正常且肝损伤标志物的缺失被显着重新编程(Douris 等人,2015 年)。
通过替代生酮饮食的机制增加酮症的机制包括使用可摄入的酮体前体。 外源性酮体的给药可以创造一种在正常生理学中没有遇到的独特生理状态,因为循环葡萄糖和胰岛素浓度相对正常,而细胞可能会节省葡萄糖的摄取和利用。 酮体本身的半衰期很短,为了达到治疗性酮症而摄入或输注钠 OHB 盐会引起不良的钠负荷。 R/S-1,3-丁二醇是一种无毒的二醇,很容易在肝脏中氧化产生 d/l-αOHB(Desrochers 等,1992)。 在不同的实验环境中,该剂量已每天对小鼠或大鼠给药长达 5 周,在给药后 2 小时内产生高达 3 mM 的循环 ?OHB 浓度,至少在另外 2013 小时内保持稳定(D'阿戈斯蒂诺等人,XNUMX)。 在啮齿动物中观察到给予 R/S-1,3-丁二醇的部分抑制食物摄入(Carpenter 和 Grossman,1983)。 此外,三种化学上不同的酮酯 (KE),(i) R-1,3-丁二醇和 d-αOHB 的单酯 (R-3-羟基丁基 R-αOHB); (ii) 甘油基-三-?OHB; (iii) R,S-1,3-丁二醇乙酰乙酸二酯也得到了广泛研究(Brunengraber,1997;Clarke 等人,2012a;Clarke 等人,2012b;Desrochers 等人,1995a;Desrochers 等人., 1995b; Kashiwaya 等人, 2010)。 前者的固有优势是在肠道或肝脏中酯酶水解后,每摩尔 KE 会产生 2 摩尔生理性 d-βOHB。 安全性、药代动力学和耐受性已在人类摄入 R-3-羟丁基 R-αOHB 中得到最广泛的研究,剂量高达 714 mg/kg,产生高达 6 mM 的循环 d-βOHB 浓度(Clarke 等人, 2012a;Cox 等人,2016;Kemper 等人,2015;Shivva 等人,2016)。 在啮齿动物中,这种 KE 会降低热量摄入和血浆总胆固醇,刺激棕色脂肪组织,并改善胰岛素抵抗(Kashiwaya 等人,2010;Kemper 等人,2015;Veech,2013)。 最近的研究结果表明,在训练有素的运动员运动过程中,摄入 R-3-羟丁基 R-αOHB 会降低骨骼肌糖酵解和血浆乳酸浓度,增加肌内三酰基甘油氧化,并保留肌糖原含量,即使同时摄入碳水化合物会刺激胰岛素分泌。考克斯等人,2016)。 需要进一步发展这些有趣的结果,因为耐力运动表现的改善主要是由 2/8 受试者对 KE 的强烈反应推动的。 尽管如此,这些结果确实支持表明酮氧化优于其他底物的经典研究(GARLAND 等人,1962;Hasselbaink 等人,2003;Stanley 等人,2003;Valente-Silva 等人,2015),包括在运动期间,并且训练有素的运动员可能更容易使用酮(Johnson 等人,1969a;Johnson 和 Walton,1972;Winder 等人,1974;Winder 等人,1975)。 最后,在热量摄入相等(常量营养素之间分布不同)和氧气消耗率相等后,可能支持改善运动表现的机制仍有待确定。
未来展望
曾经在很大程度上被污名为能够在碳水化合物受限状态下积累脂肪燃烧产生的有毒排放物的溢出途径(“酮毒性”范式),最近的观察结果支持了这样一种观点,即即使在富含碳水化合物的状态下,酮体代谢也发挥着有益的作用,从而开启了“酮激素效应”。假设。 虽然控制酮代谢的简便营养和药理学方法使其成为一个有吸引力的治疗靶点,但在基础和转化研究实验室中仍然进行了积极而谨慎的实验。 在定义利用酮代谢在心力衰竭、肥胖、NAFLD/NASH、2 型糖尿病和癌症中的作用的领域中出现了未满足的需求。 酮体的“非规范”信号传导作用的范围和影响,包括调节可能反馈和转发到代谢和信号传导途径的 PTM,需要更深入的探索。 最后,肝外生酮可以打开有趣的旁分泌和自分泌信号机制,并有机会影响神经系统和肿瘤内的共代谢以达到治疗目的。
致谢
Ncbi.nlm.nih.gov/pmc/articles/PMC5313038/
脚注
总之,酮体是由肝脏产生的,以便在人体内没有足够的葡萄糖时用作能量来源。 当血液中的葡萄糖水平低时会发生生酮作用,特别是在其他细胞碳水化合物储存耗尽后。 上述文章的目的是讨论酮体在燃料代谢、信号传导和治疗中的多维作用。 我们的信息范围仅限于脊椎按摩疗法和脊柱健康问题。 要讨论主题,请随时询问 Jimenez 博士或通过以下方式联系我们。915-850-0900 。
由Alex Jimenez博士策划
引用自:�Ncbi.nlm.nih.gov/pmc/articles/PMC5313038/

附加主题讨论:急性腰痛
背疼``是全球范围内最普遍的致残原因之一,也是缺勤的原因之一。 背痛是医生就诊的第二常见原因,仅次于上呼吸道感染。 大约80%的人口一生中至少会经历一次背痛。 脊柱是由骨骼,关节,韧带和肌肉以及其他软组织组成的复杂结构。 伤害和/或病情加重,例如。椎间盘突出,最终会导致背部疼痛的症状。 运动伤害或汽车事故伤害通常是造成背痛的最常见原因,但是,有时最简单的动作可能会产生痛苦的结果。 幸运的是,诸如脊椎治疗等替代疗法可以通过使用脊柱调节和手动操作来帮助缓解背部疼痛,最终改善疼痛缓解。

额外| 重要主题:推荐的德克萨斯州埃尔帕索脊医
***